Anomalies de l’intervalle QT
L’espace QT (qui mesure le temps du début du QRS à la fin de l’onde T) doit être corrigé en fonction de la fréquence cardiaque, du sexe et accessoirement de l’âge du sujet, selon une formule ou des tables qui se trouvent au chapitre 1.
L’anomalie la plus importante de l’espace QTc est son prolongement, qui provoque une inhomogénéité de la repolarisation avec une tendance marquée à induire des troubles du rythme ventriculaire graves (torsades de pointe). Ce prolongement de l’espace QT se retrouve dans plusieurs situations cliniques citées ci-dessous:
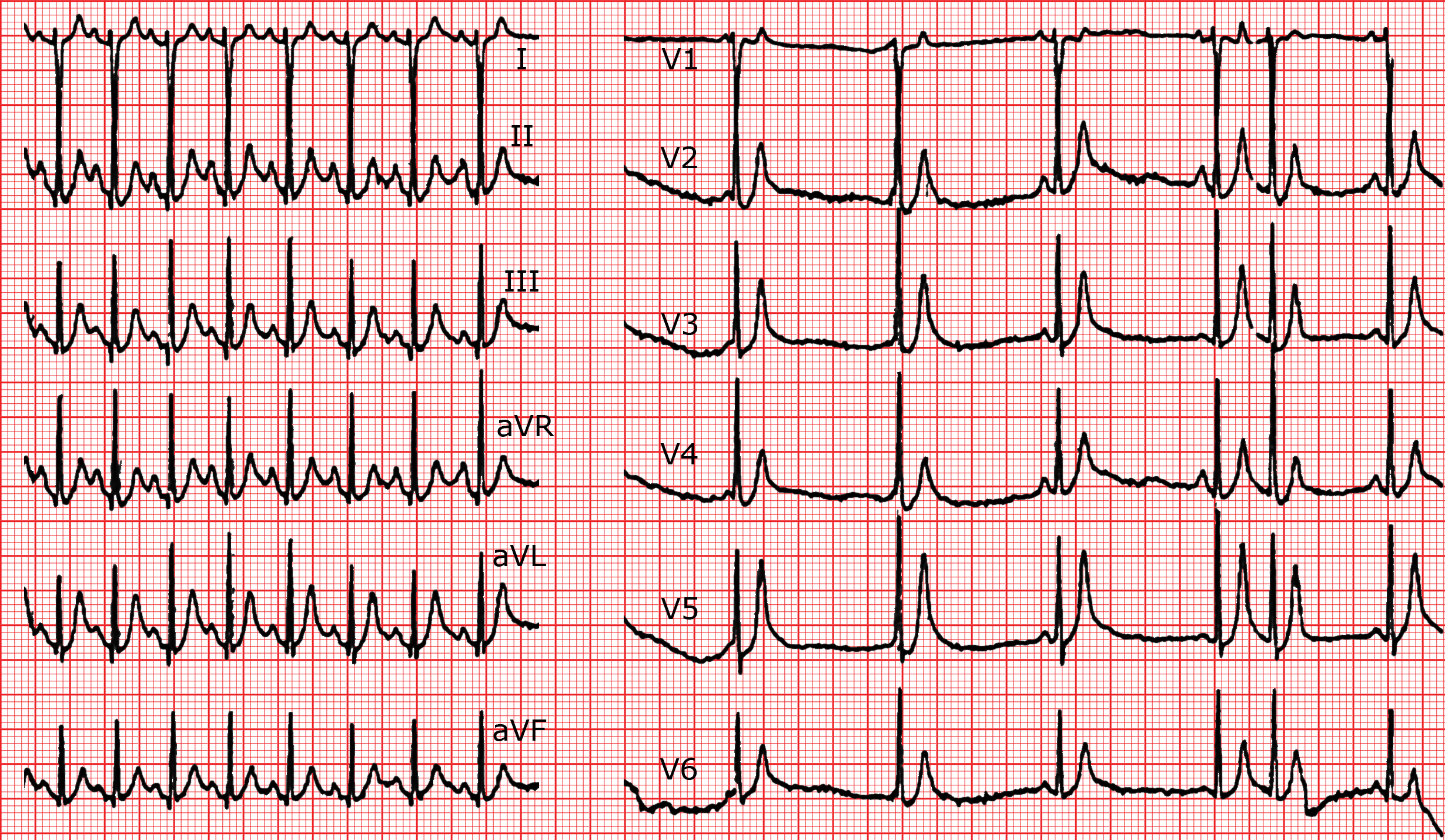
A) Syndrome du QT long congénital
Il s’agit d’une affection familiale héréditaire très rare, appelée syndrome de Romano-Ward lorsque l’anomalie est isolée, ou de syndrome de Jervell-Lange-Nielsen quand elle est associée à une surdité. Le prolongement de l’espace QT est à l’origine de torsades de pointe, favorisées par le stress, l’émotion ou l’exercice physique, susceptibles de dégénérer en fibrillation ventriculaire. Plusieurs mutations génétiques ont été décrites en association avec ce syndrome. Unesyncope est souvent le premier symptôme de la maladie. De manière générale il faut toujours prendre très au sérieux les syncopes qui surviennent, surtout à l’effort, chez l’enfant ou le jeune adolescent.
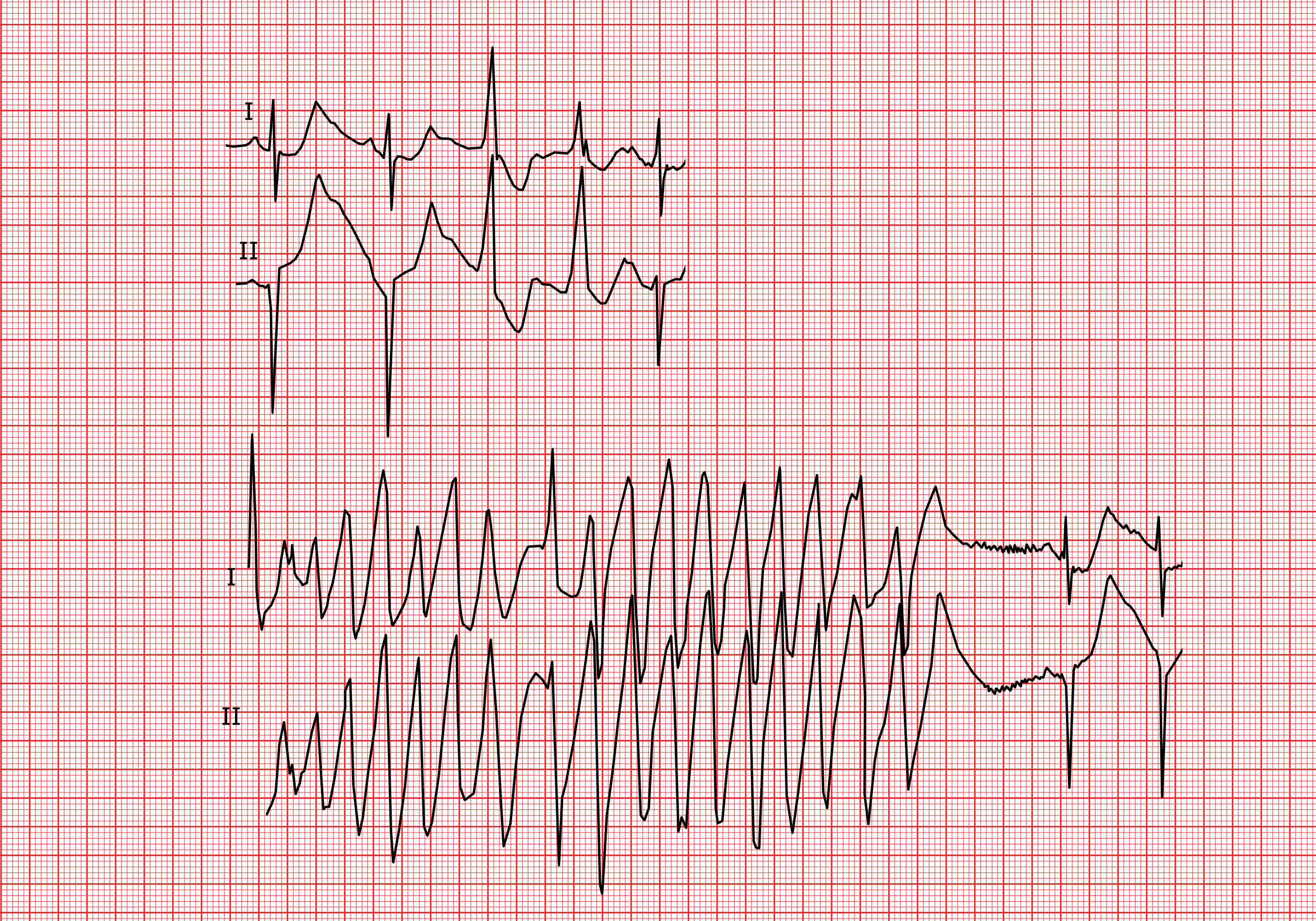
B) Syndrome du QT long acquis
Le syndrome du QT long acquis est le plus souvent iatrogène ou associé aux pathologies suivantes:
-
Ischémie.
-
Hémorragie sous-arachnoïdienne.
-
Dysthyroïdie.
-
Troubles électrolytiques (hypocalcémie).
-
Effets secondaires médicamenteux lors d’un traitement avec des antiarythmiques (particulièrement ceux de la classe IA, du type quinidine, ou de la classe III tels le sotalol ou l’amiodarone), des antidépresseurs, certains anti-histaminiques ou encore d’autres substances. Des listes exhaustives sont disponibles dans la littérature.
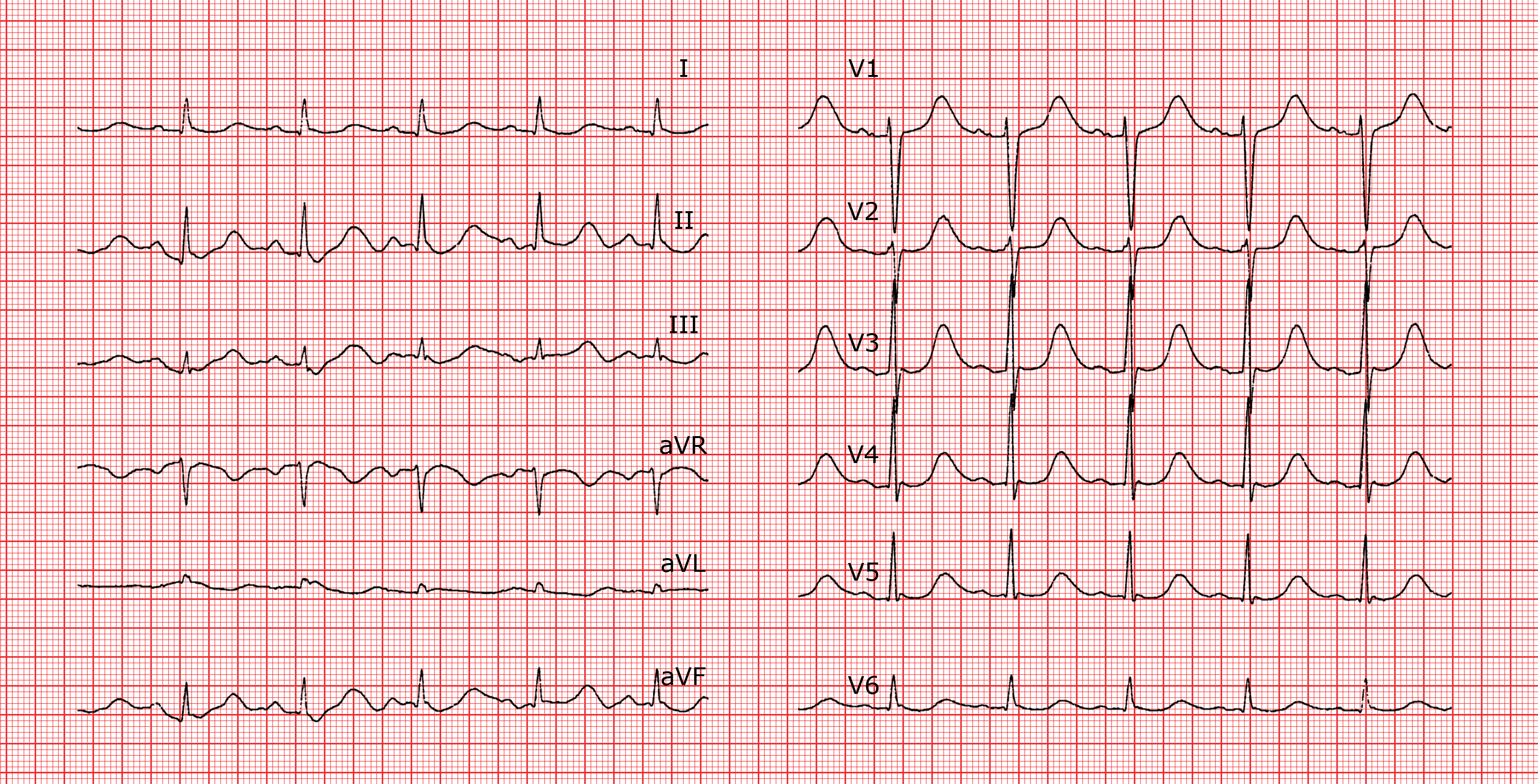
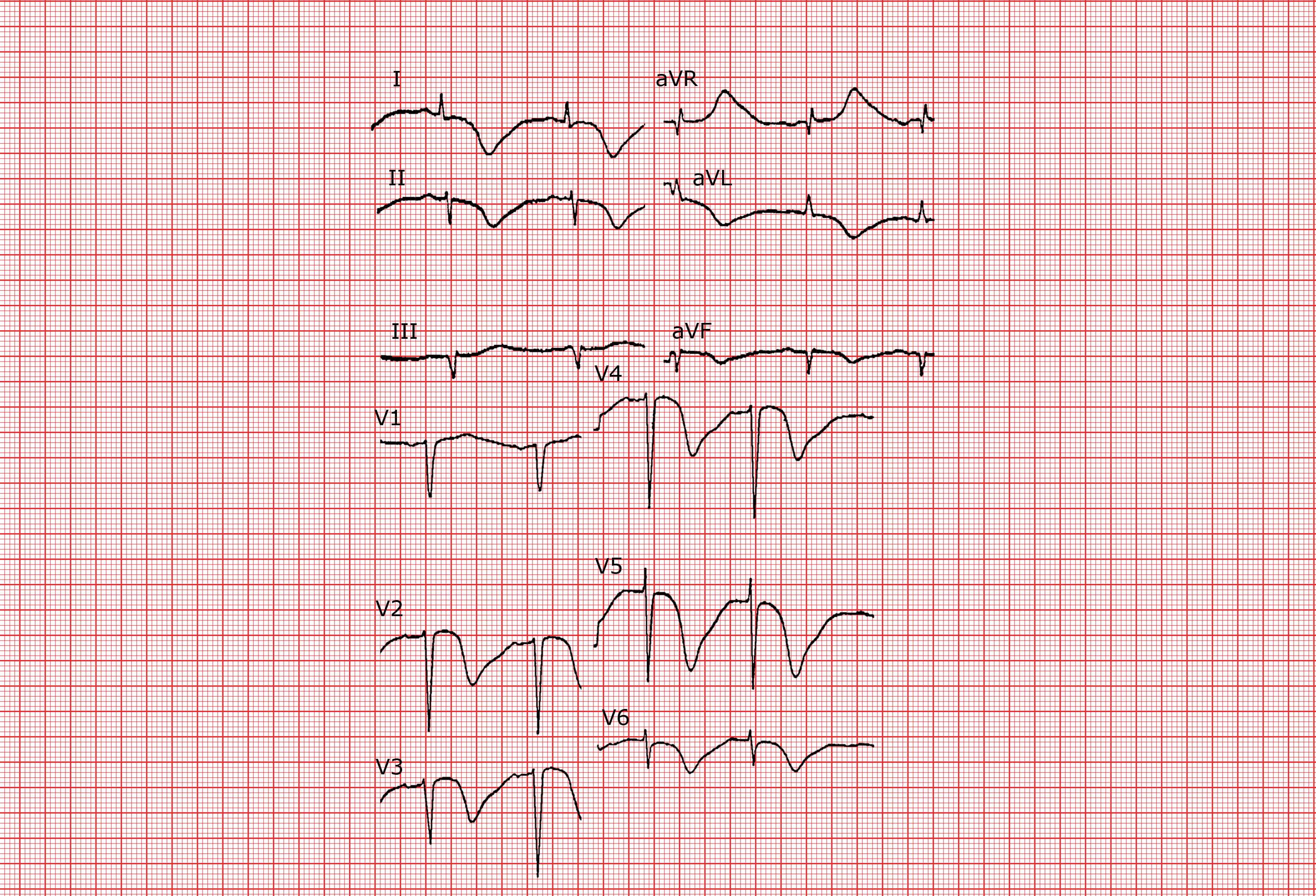
Syndrome du QT court
Le syndrome du QT court congénital est défini par un intervalle QTc ≤ 340 ms. Il peut être responsable de tachyarythmies ventriculaires et/ou auriculaires. Plusieurs mutations génétiques ont été décrites aboutissant à un raccourcissement du potentiel d’action par anomalie des canaux potassiques. Ce syndrome est associé à un risque élevé de mort subite. L’intervalle QT n’est que peu modifié par la fréquence cardiaque d’où la difficulté à poser le diagnostic à des fréquences rapides (en particulier chez l’enfant) lorsque le segment ST est très court voire absent. L’onde T est souvent grande, pointue et symétrique. L’intervalle PR peut parfois être sous-décalé. A l’examen électrophysiologique on induit facilement une fibrillation ventriculaire mais aussi une fibrillation auriculaire, car les périodes réfractaires sont très courtes.